
Single Colony Isolation


Robert Koch, in spelling out the criteria for demonstrating the etiologic agent for a disease, emphasized as two of his crucial criteria that one must isolate the putative agent in pure culture, and then experimentally cause the disease in a healthy animal with this pure culture.
The technique which he developed to produce a pure culture, the idea for which came from his observation of colonies growing on the spoiling half of a baked potato, is termed single colony isolation. A colony is picked with a sterile loop, streaked out on a sterile plate. The loop is resterilized, and the initial streak is spread out across the plate in successive cycles of sterilization and cross streaking. The goal is to sufficiently spread out the inoculum so that individual colonies are formed. For rigorous applications, this process should be performed twice in succession. Practice the fluid movements of streaking (6, 7 & 8) with a pencil on paper to perfect the proper motion.
Make a careful full-sized plate illustration in your lab book practicing the primary, secondary and tertiary streaks as shown by the instructor. Always flame and cool the loop between each streaking. Make detailed notes in your book regarding the origin of the mixed culture, and the lineage of the colonies selected.

PREPARE SOURCE PLATE: Spread a suitably diluted mixed culture on a plate to give 100 to 500 colonies per plate, incubate until well-formed colonies appear (24-48 hours). [For example, from coliform plate count on EMB lac plates] This is your source plate.

SCORE SOURCE COLONIES: Examine the source plate under a magnifying glass. How many classes can you identify? Pick colonies to be isolated: one lactose fermenting colony, and three other highly distinct non-lactose-fermenting colonies.

Select, circle and number them with a wax pencil: four colonies as distinctly different from each other as possible, the first of which is lactose positive, the latter three unambiguously lactose non-fermenting.

Prepare a table to record the traits of these four selected colonies, as shown. in your notebook: size, morphology, color, wrinkled, mucoid, spreading, clear, etc.

PREPARE THE STREAK PLATE: Mark a fresh agar plate of the same type as in step 1 with a wax pencil to divide it in fourths. In small print, enter your initials, seat number and the date at one edge and label the four areas with the coded numbers of the selected colonies.

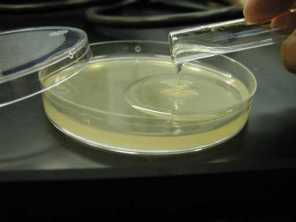
PREPARE THE LOOP FOR OPTIMUM PERFORMANCE: After flaming to ensure sterility, straighten out the shaft of the loop, ensure a smooth loop at the end, and fashion an angle towards the end to make picking a colony more precise.

STERILIZE ADJUST LOOP : Flame a bacteriological loop so that the entire wire glows. When cool, adjust the 26 gauge platinum wire to the proper shape: 4 cm straight shank, the last 6 mm bent at a slight angle, tipped with a 1-2 mm loop, at a right angle to axis of handle.

PICK THE COLONY: Reflame the loop. Open the source plate, touch the hot loop to a sterile part of the agar to insure that it is cool. Lightly touch the edge of the first selected colony so that you pick up a small sample. (If you pick up too much, it will not streak out properly. If you pick up too little, the transfer may not successful.) Close the source plate.

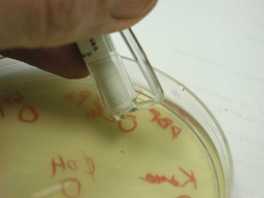
APPLY PRIMARY STREAK: On the new plate, with reflected light showing your tracks, apply the primary streak by lightly zig-zagging the inoculating loop across the agar at one side of the semicircle with a small wiggling/dragging motion. Do not press hard enough to dig into the agar, nor to bend the loop.

PERFORM SECONDARY STREAK: Reflame and cool the loop as before. With a light sweeping motion (wrist stiff, pivot from elbow, little finger resting on table surface), perform the secondary streak: start at the terminal end of the primary streak, and sweep across the top of the semicircle, cross streaking several times back and forth through the primary streak.

MAKE TERTIARY STREAK: Reflame and cool the loop as before. Rotate the plate 90 degrees, and cross streak through the secondary streak, starting at the far end, working towards the primary streak, and filling the rest of the semicircle. Do not go through the primary streak.

For the remaining colonies to be purified, repeat the streaking procedure (steps 5 through 8). Here is the finished plate with four specimens streaked out, prior to incubation.

Incubate for 24-48 hours at 37 C or until colonies are well formed. (Do not “overgrow.”) Score your streaking abilities: do you see numerous individual colonies well separated? Great! Note that the primary streak is confluent. Score the morphologies of the two isolated strains. Are they identical with the original colonies from step 2? Record your results.

Here is an EMB plate streaked with a mixed culture of E. coli and Salmonella typhimurium (the streaks on the left and on the right). Perfect single colony isolation produces individual purple (E. coli) and pink (S. typhimurium) colonies. The streak at the top is pure E. coli. The streak at the bottom did not grow…
GRADING OF STREAKING ABILITY:
Prepare mixed culture of E coli and S typhimurium. Use also as sources of bacteria TSI slants with these bacteria on their surfaces (or colonies of each). Have student mark plate into fourths, with Ec and St across from each other, the mixed culture streaked in two quadrants across from each other as well. Tell them to mark the plates with the usual information (initials, date, seat, and the quadrants marked, and to enter the data in their notebooks.
Grade the plates as follows:
Proper technique:
initials
date
seat
streaks:
not touching or cross contaminated
single colonies in each streak get 2 points, including both St and Ec in the mixed culture streaks.
Total score: 15 points (3 for technique, 2 for Ec, 2 for St, 2+2 for each of the two mixed cultures.















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}